|
|
Post by Life on Apr 9, 2020 20:58:05 GMT 5
Scientific assessment suggest horseshoe bats being the chief carriers and transmitters of coronaviruses worldwide. Recommended studies:- Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemicAbstractThere are outstanding evolutionary questions on the recent emergence of coronavirus SARS-CoV-2/hCoV-19 in Hubei province that caused the COVID-19 pandemic, including (1) the relationship of the new virus to the SARS-related coronaviruses, (2) the role of bats as a reservoir species, (3) the potential role of other mammals in the emergence event, and (4) the role of recombination in viral emergence. Here, we address these questions and find that the sarbecoviruses – the viral subgenus responsible for the emergence of SARS-CoV and SARS-CoV-2 – exhibit frequent recombination, but the SARS-CoV-2 lineage itself is not a recombinant of any viruses detected to date. In order to employ phylogenetic methods to date the divergence events between SARS-CoV-2 and the bat sarbecovirus reservoir, recombinant regions of a 68-genome sarbecovirus alignment were removed with three independent methods. Bayesian evolutionary rate and divergence date estimates were consistent for all three recombination-free alignments and robust to two different prior specifications based on HCoV-OC43 and MERS-CoV evolutionary rates. Divergence dates between SARS-CoV-2 and the bat sarbecovirus reservoir were estimated as 1948 (95% HPD: 1879-1999), 1969 (95% HPD: 1930-2000), and 1982 (95% HPD: 1948-2009). Despite intensified characterization of sarbecoviruses since SARS, the lineage giving rise to SARS-CoV-2 has been circulating unnoticed for decades in bats and been transmitted to other hosts such as pangolins. The occurrence of a third significant coronavirus emergence in 17 years together with the high prevalence and virus diversity in bats implies that these viruses are likely to cross species boundaries again. In Brief The Betacoronavirus SARS-CoV-2 is a member of the sarbecovirus subgenus which shows frequent recombination in its evolutionary history. We characterize the extent of this genetic exchange and identify non-recombining regions of the sarbecovirus genome using three independent methods to remove the effects of recombination. Using these non-recombining genome regions and prior information on coronavirus evolutionary rates, we obtain estimates from three approaches that the most likely divergence date of SARS-CoV-2 from its most closely related available bat sequences ranges from 1948 to 1982. Key Points- RaTG13 is the closest available bat virus to SARS-CoV-2; a sub-lineage of these bat viruses is able to infect humans. Two sister lineages of the RaTG13/SARS-CoV-2 lineage infect Malayan pangolins.
- The sarbecoviruses show a pattern of deep recombination events, indicating that there are high levels of co-infection in horseshoe bats and that the viral pool can generate novel allele combinations and substantial genetic diversity; the sarbecoviruses are efficient ‘explorers’ of phenotype space.
- The SARS-CoV-2 lineage is not a recent recombinant, at least not involving any of the bat or pangolin viruses sampled to date.
- Non-recombinant regions of the sarbecoviruses can be identified, allowing for phylogenetic inference and dating to be performed. We constructed three such regions using different methods.
- We estimate that RaTG13 and SARS-CoV-2 diverged 40 to 70 years ago. There is a diverse unsampled reservoir of generalist viruses established in horseshoe bats.
- While an intermediate host responsible for the zoonotic event cannot be ruled out, the relevant evolution for spillover to humans very likely occurred in horseshoe bats.
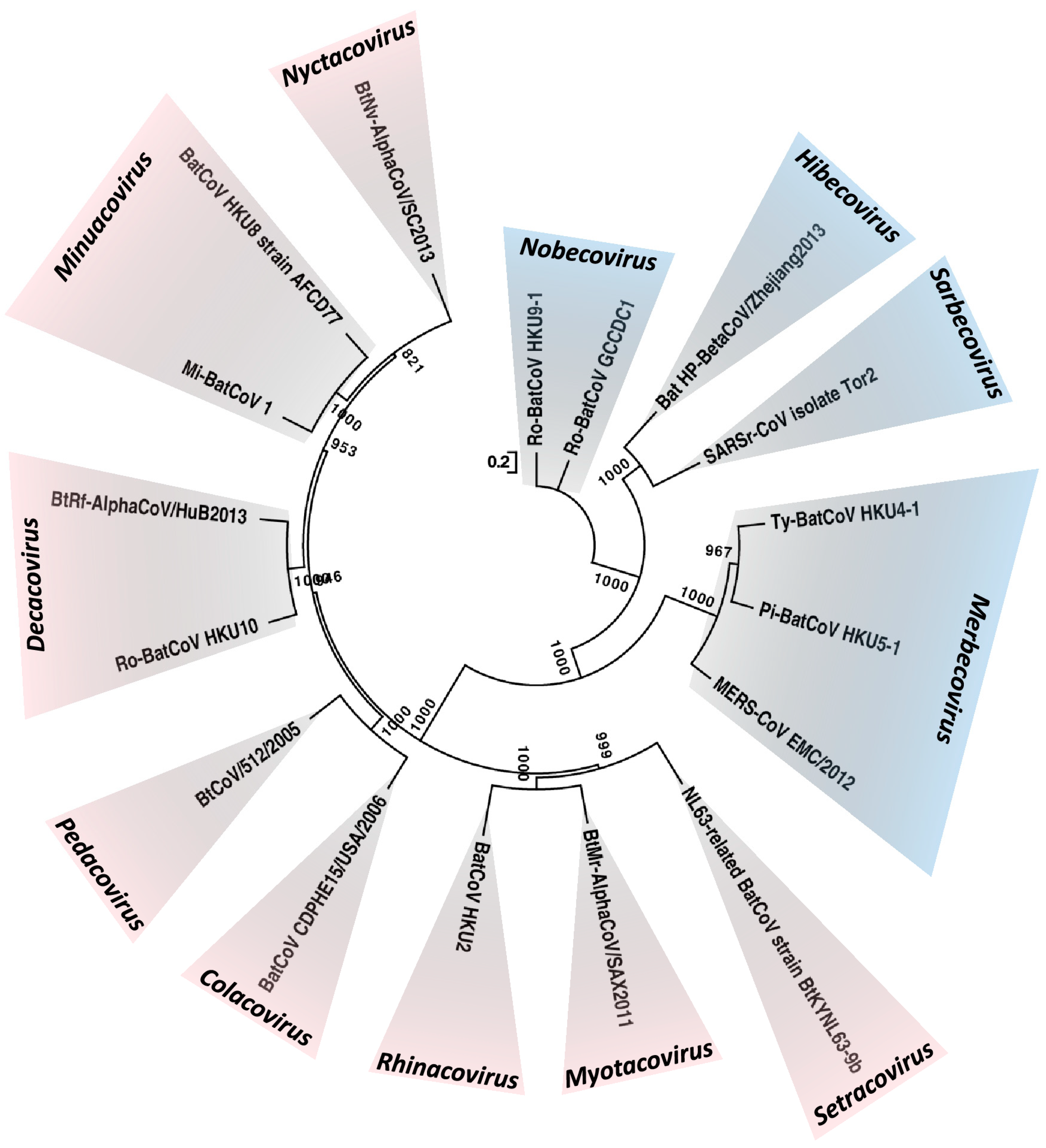
--- Global Epidemiology of Bat Coronaviruses Abstract Bats are a unique group of mammals of the order Chiroptera. They are highly diversified and are the group of mammals with the second largest number of species. Such highly diversified cell types and receptors facilitate them to be potential hosts of a large variety of viruses. Bats are the only group of mammals capable of sustained flight, which enables them to disseminate the viruses they harbor and enhance the chance of interspecies transmission. This article aims at reviewing the various aspects of the global epidemiology of bat coronaviruses (CoVs). Before the SARS epidemic, bats were not known to be hosts for CoVs. In the last 15 years, bats have been found to be hosts of >30 CoVs with complete genomes sequenced, and many more if those without genome sequences are included. Among the four CoV genera, only alphaCoVs and betaCoVs have been found in bats. As a whole, both alphaCoVs and betaCoVs have been detected from bats in Asia, Europe, Africa, North and South America and Australasia; but alphaCoVs seem to be more widespread than betaCoVs, and their detection rate is also higher. For betaCoVs, only those from subgenera Sarbecovirus, Merbecovirus, Nobecovirus and Hibecovirus have been detected in bats. Most notably, horseshoe bats are the reservoir of SARS-CoV, and several betaCoVs from subgenus Merbecovirus are closely related to MERS-CoV. In addition to the interactions among various bat species themselves, bat–animal and bat–human interactions, such as the presence of live bats in wildlife wet markets and restaurants in Southern China, are important for interspecies transmission of CoVs and may lead to devastating global outbreaks. Images Figure 1. Maximum-likelihood phylogeny based on the complete genome sequences of 17 bat CoV species released by ICTV in 2018. A general time-reversible model of nucleotide substitution with estimated base frequencies, the proportion of invariant sites, and the γ distribution of rates across sites were used in the maximum-likelihood analysis. Bootstrap values are shown next to the branches. The scale bar indicates the number of nucleotide substitutions per site. Different colors represent different genera. Red, Alphacoronavirus; blue, Betacoronavirus. Updated subgenera clusters are labelled Setracovirus, Myotacovirus, Rhinacovirus, Colacovirus, Pedacovirus, Decacovirus, Minunacovirus, Nyctacovirus for the Alphacoronavirus and Nobecovirus, Hibecovirus, Sarbecovirus, Merbecovirus for the Betacoronavirus.
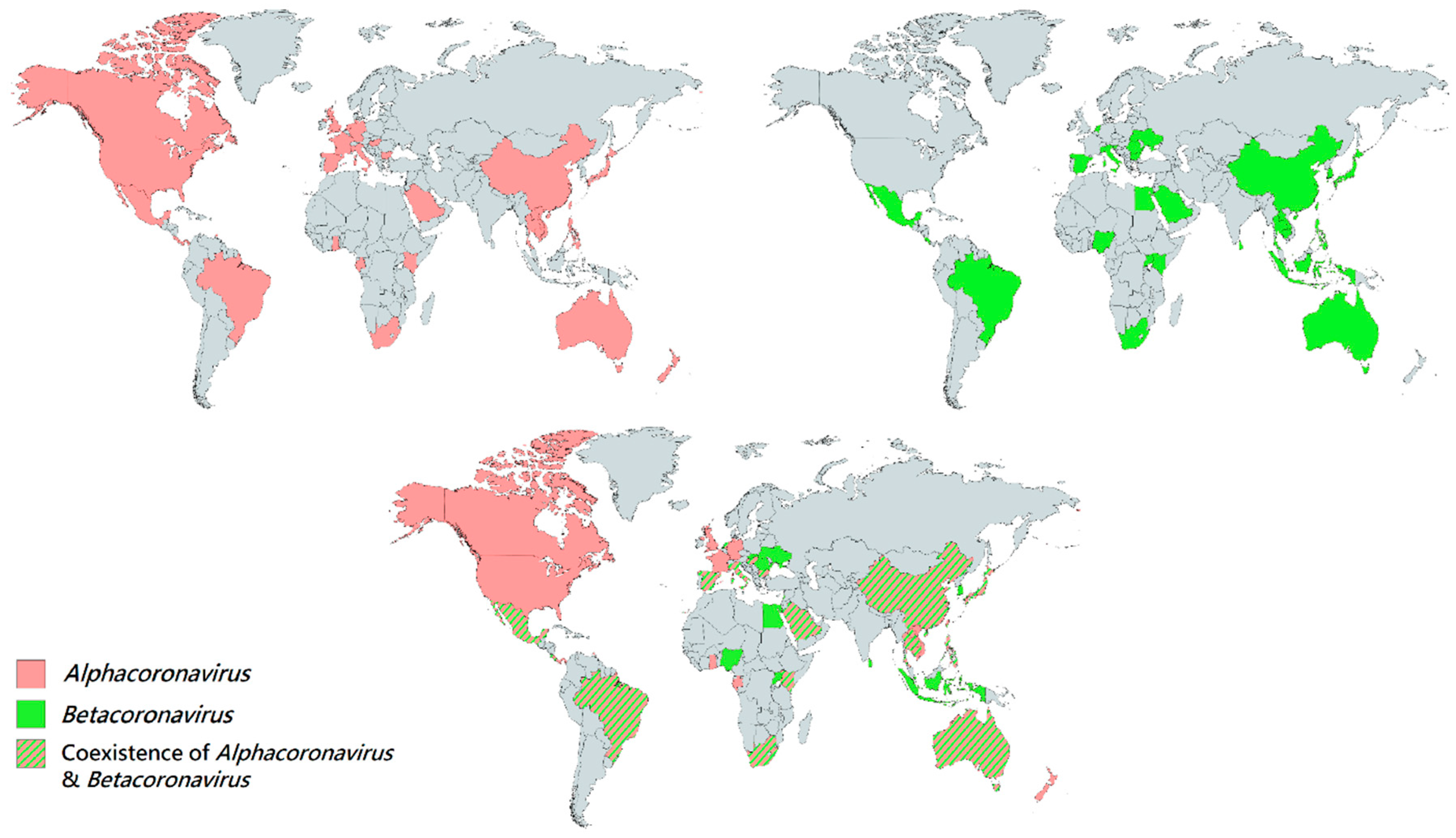 Figure 2. Geographical distribution of bat CoVs from the genera Alphacoronavirus and Betacoronavirus. Each colored region represents the country which reported the discovery of bat CoV. Red regions represent the countries which discovered bat Alphacoornavirus. Green regions represent the countries which discovered bat Betacoronavirus. Red-green striped regions represent the countries which discovered both bat Alphacoronavirus and Betacoronavirus.
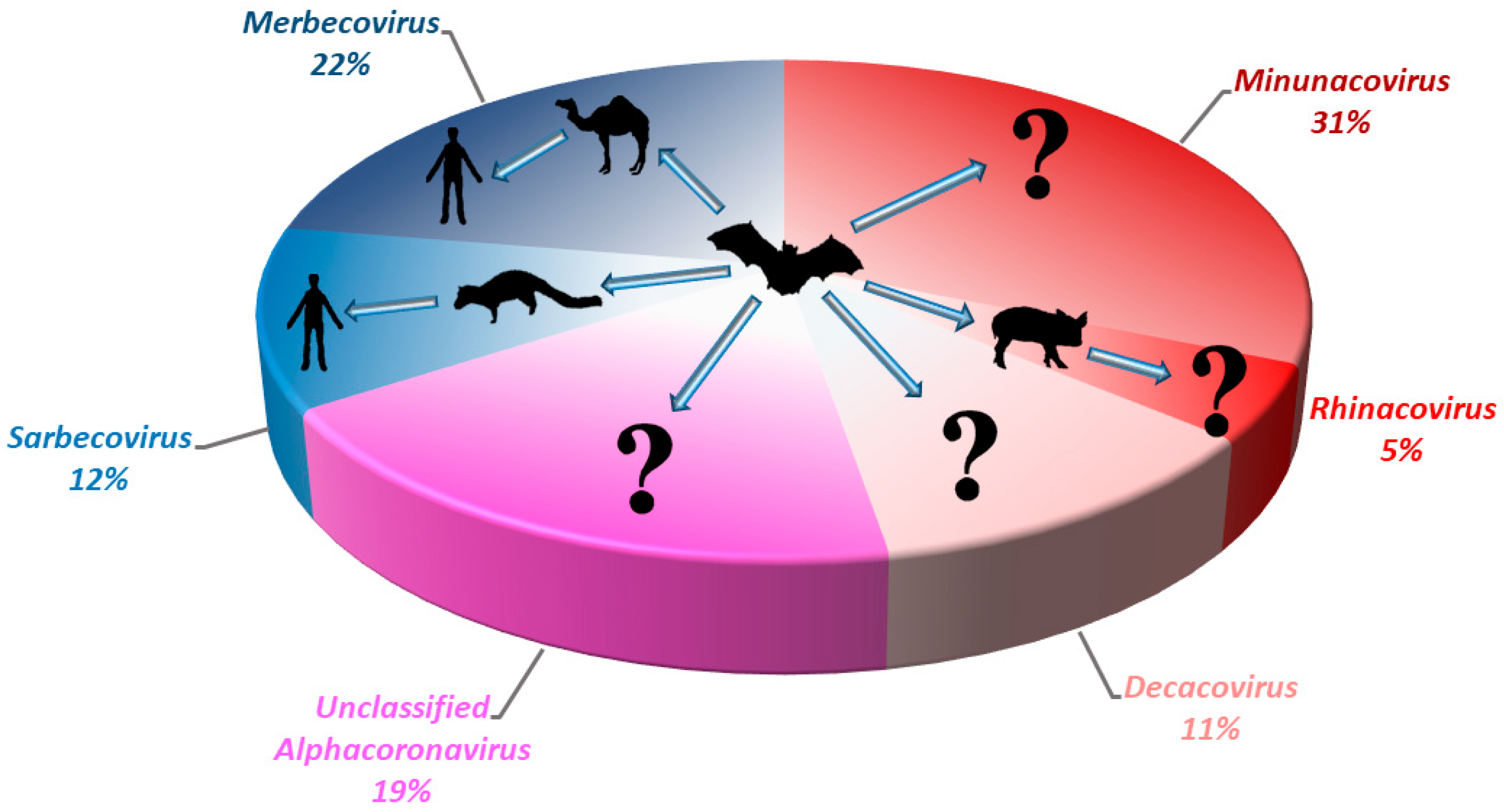
Figure 3. Pie chart showing the relative detection rate of different bat CoVs from different subgenera of Alphacoronavirus and Betacoronavirus in Hong Kong from 2008 to 2017. The potential zoonotic transmission routes of each sub-genus of bat CoV detected are shown. Unclassified Alphacoronavirus represents those without complete genome sequences or genome characterization. Red color represents the sub-genera from Alphacoronavirus; Blue color represents the sub-genera from Betacoronavirus.
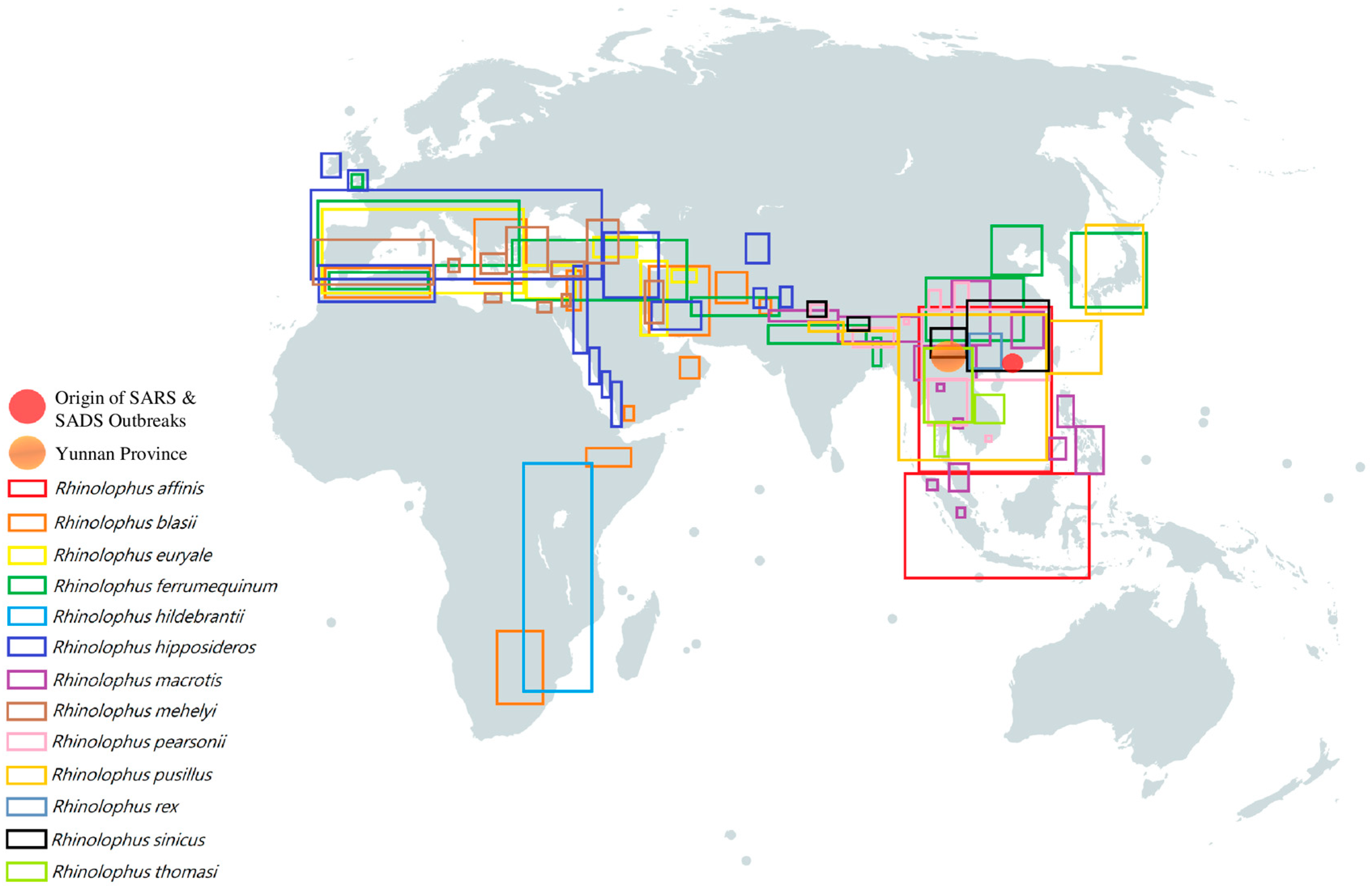 Figure 4. Geographical distribution of different horseshoe bats which were discovered to carry SARS-like BatCoV [114,115,116,117,118,119,120,121,122,123,124,125]. Each colored rectangular box represents the geographical distribution of a specific horseshoe bat species respectively: red box, Rhinolophus affinis; orange box, Rhinolophus blasii; yellow box, Rhinolophus euryale; green box, Rhinolophus ferrumequinum; turquoise box, Rhinolophus hildebrantii; indigo box, Rhinolophus hipposideros; purple box, Rhinolophus macrotis; brown box, Rhinolophus mehelyi; pink box, Rhinolophus pearsonii; gold box, Rhinolophus pusillus; blue-gray box, Rhinolophus rex; black box, Rhinolophus sinicus; lime box, Rhinolophus thomasi. Orange circle represents Yunnan Province; Red circle represents the origin of SARS & SADS outbreaks.
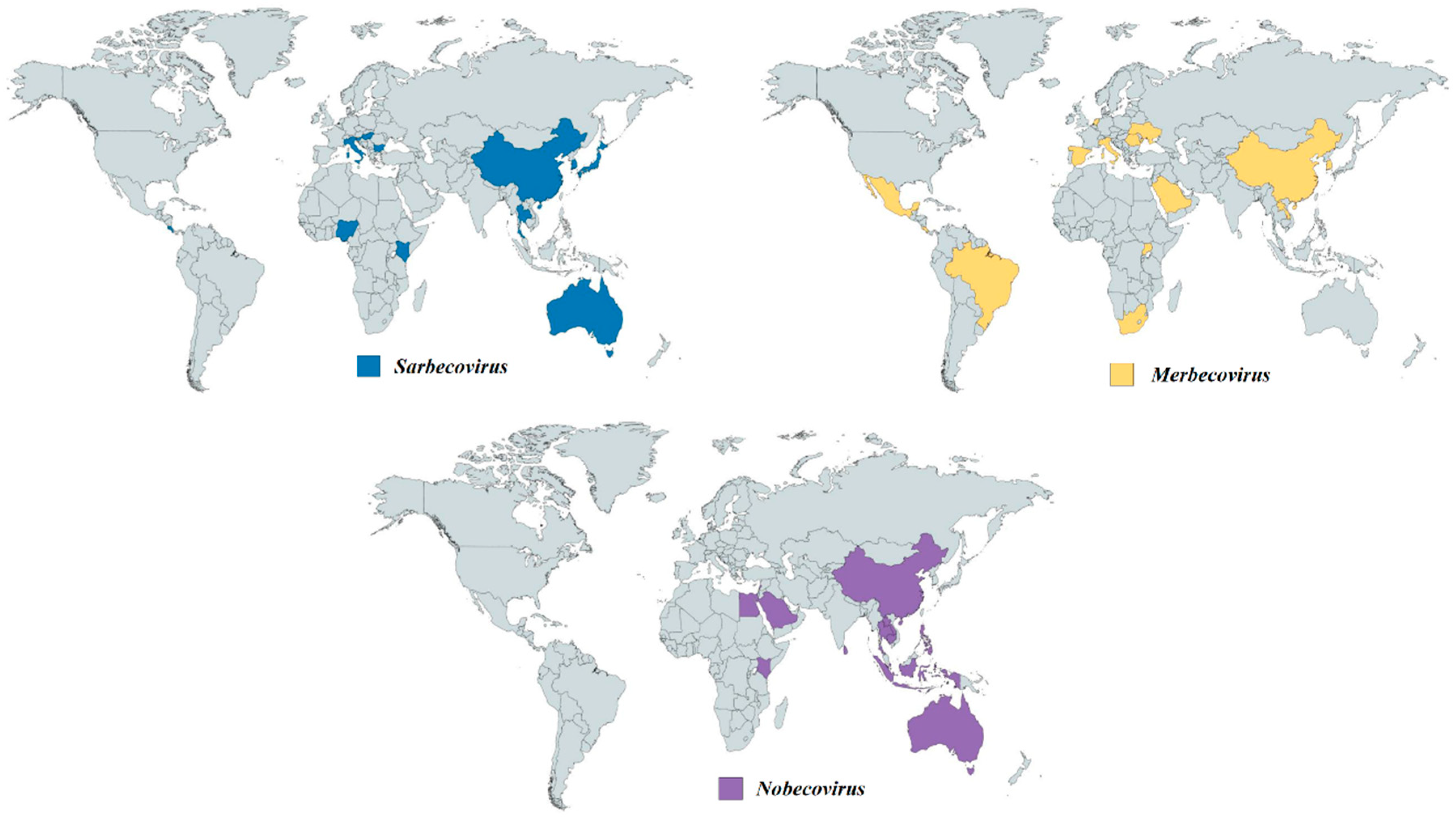 Figure 5. Geographical distribution of bat CoVs from the genus Betacoronavirus. Each colored region represents the country which reported the discovery of bat CoV from different sub-genera. Navy-blue regions represent the countries which discovered bat CoVs from Sarbecovirus. Yellow regions represent the countries which discovered bat CoVs from Merbecovirus. Purple regions represent the countries which discovered bat CoVs from Nobecovirus.
|
|
|
|
Post by Life on Apr 10, 2020 14:17:00 GMT 5
Fig. 1: Features of the spike protein in human SARS-CoV-2 and related coronaviruses. 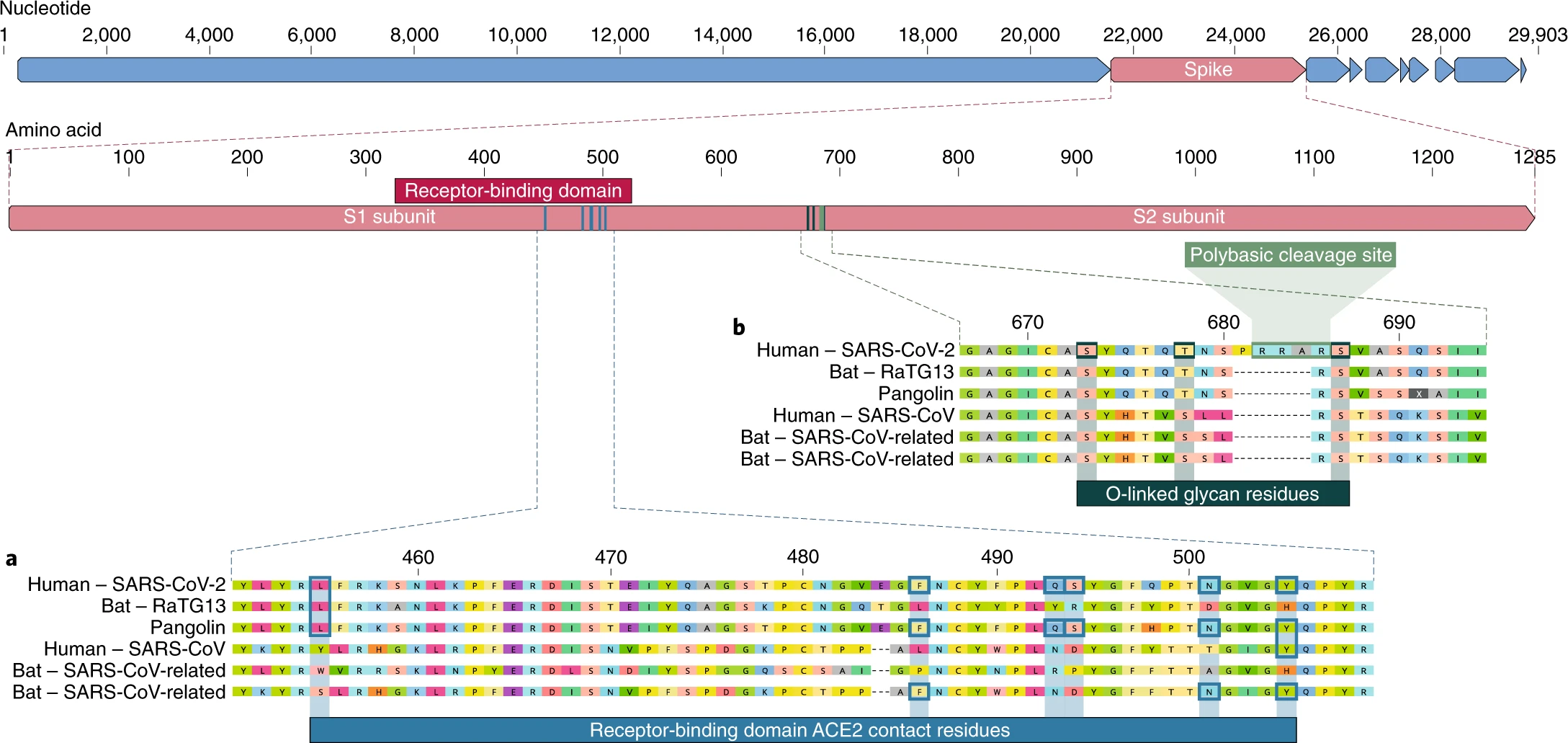 a, a, Mutations in contact residues of the SARS-CoV-2 spike protein. The spike protein of SARS-CoV-2 (red bar at top) was aligned against the most closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues in the spike protein that make contact to the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in lineage B betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146 and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378 (NCBI BioProject PRJNA573298) 29,30. Taken from the following study: The proximal origin of SARS-CoV-2AbstractSince the first reports of novel pneumonia (COVID-19) in Wuhan, Hubei province, China 1,2, there has been considerable discussion on the origin of the causative virus, SARS-CoV-2 3 (also referred to as HCoV-19) 4. Infections with SARS-CoV-2 are now widespread, and as of 11 March 2020, 121,564 cases have been confirmed in more than 110 countries, with 4,373 deaths 5. SARS-CoV-2 is the seventh coronavirus known to infect humans; SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease, whereas HKU1, NL63, OC43 and 229E are associated with mild symptoms6. Here we review what can be deduced about the origin of SARS-CoV-2 from comparative analysis of genomic data. We offer a perspective on the notable features of the SARS-CoV-2 genome and discuss scenarios by which they could have arisen. Our analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus. Full read: www.nature.com/articles/s41591-020-0820-9
|
|
|
|
Post by creature386 on Apr 10, 2020 16:56:19 GMT 5
|
|
|
|
Post by Life on Apr 11, 2020 11:12:42 GMT 5
Below is a limited view of the types of coronavirues which are assumed to have caused mild symptoms in humans but spread at an earlier point in time ( HKU1, NL63, OC43 and 229E respectively). --- Human Coronavirus Circulation in the United States 2014-2017AbstractBackground: Human coronaviruses (HCoVs) -OC43, -229E, -NL63 and -HKU1 cause upper and lower respiratory tract infections. HCoVs are globally distributed and the predominant species may vary by region or year. Prior studies have shown seasonal patterns of HCoV species and annual variation in species prevalence but national circulation patterns in the US have not yet been described. Objectives: To describe circulation patterns of HCoVs -OC43, -229E, -NL63 and -HKU1 in the US. Study design: We reviewed real-time reverse transcription polymerase chain reaction (rRT-PCR) test results for HCoV-OC43, -229E, -NL63 and -HKU1 reported to The National Respiratory and Enteric Virus Surveillance System (NREVSS) by U.S. laboratories from July 2014-June 2017. We calculated the total number of tests and percent positive by week. For a subset of HCoV positive submissions with age and sex of the patient available, we tested for differences in age and sex across the four HCoV species using Chi Square and Kruskal Wallace tests. Results: 117 laboratories reported 854,575 HCoV tests; 2.2% were positive for HCoV-OC43, 1.0% for HCoV-NL63, 0.8% for HCoV-229E, and 0.6% for HCoV-HKU1. The percentage of positive tests peaked during December - March each year. No significant differences in sex were seen across species, although a significant difference in age distribution was noted. Conclusions: Common HCoVs may have annual peaks of circulation in winter months in the US, and individual HCoVs may show variable circulation from year to year. Different HCoV species may be detected more frequently in different age groups. Further years of data are needed to better understand patterns of activity for HCoVs. Full read: pubmed.ncbi.nlm.nih.gov/29427907/?from_term=coronavirus&from_pos=6--- Human Coronavirus-Associated Influenza-Like Illness in the Community Setting in PeruAbstractWe present findings describing the epidemiology of non-severe acute respiratory syndrome human coronavirus-associated influenza-like illness from a population-based active follow-up study in four different regions of Peru. In 2010, the prevalence of infections by human coronaviruses 229E, OC43, NL63, or HKU1 was 6.4% in participants with influenza-like illness who tested negative for influenza viruses. Ten of 11 human coronavirus infections were identified in the fall–winter season. Human coronaviruses are present in different regions of Peru and are relatively frequently associated with influenza-like illness in Peru. Full read: www.ncbi.nlm.nih.gov/pmc/articles/PMC4703274/--- Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method.AbstractFour human coronaviruses ( HCoV-229E, HCoV-HKU1, HCoV-NL63, and HCoV-OC43) are associated with a range of respiratory outcomes, including bronchiolitis and pneumonia. Their epidemiologies and clinical characteristics are poorly described and are often reliant on case reports. To address these problems, we conducted a large-scale comprehensive screening for all four coronaviruses by analysis of 11,661 diagnostic respiratory samples collected in Edinburgh, United Kingdom, over 3 years between July 2006 and June 2009 using a novel four-way multiplex real-time reverse transcription-PCR (RT-PCR) assay. Coronaviruses were detected in 0.3 to 0.85% of samples in all age groups. Generally, coronaviruses displayed marked winter seasonality between the months of December and April and were not detected in summer months, which is comparable to the pattern seen with influenza viruses. HCoV-229E was the exception; detection was confined to the winter of 2008 and was sporadic in the following year. There were additional longer-term differences in detection frequencies between seasons, with HCoV-OC43 predominant in the first and third seasons and HCoV-HKU1 dominating in the second (see Results for definitions of seasons). A total of 11 to 41% of coronaviruses detected were in samples testing positive for other respiratory viruses, although clinical presentations of coronavirus monoinfections were comparable to those of viruses which have an established role in respiratory disease, such as respiratory syncytial virus, influenza virus, and parainfluenza viruses. The novel multiplex assay for real-time pan-coronavirus detection enhances respiratory virus diagnosis, overcomes potential diagnostic problems arising through seasonal variation in coronavirus frequency, and provides novel insights into the epidemiology and clinical implications of coronaviruses. Full read: www.ncbi.nlm.nih.gov/pubmed/20554810 |
|
|
|
Post by Life on Apr 11, 2020 12:36:09 GMT 5
Origin and Evolution of Pathogenic CoronavirusesAbstractSevere acute respiratory syndrome coronavirus ( SARS-CoV) and Middle East respiratory syndrome coronavirus ( MERS-CoV) are two highly transmissible and pathogenic viruses that emerged in humans at the beginning of the 21st century. Both viruses likely originated in bats, and genetically diverse coronaviruses that are related to SARS-CoV and MERS-CoV were discovered in bats worldwide. In this Review, we summarize the current knowledge on the origin and evolution of these two pathogenic coronaviruses and discuss their receptor usage; we also highlight the diversity and potential of spillover of bat-borne coronaviruses, as evidenced by the recent spillover of swine acute diarrhoea syndrome coronavirus ( SADS-CoV) to pigs.
---

Fig. 1. The genomes, genes and proteins of different coronaviruses. Coronaviruses form enveloped and spherical particles of 100–160 nm in diameter. They contain a positive-sense, single-stranded RNA (ssRNA) genome of 27–32 kb in size. The 5'-terminal two-thirds of the genome encodes a polyprotein, pp1ab, which is further cleaved into 16 non-structural proteins that are involved in genome transcription and replication. The 3' terminus encodes structural proteins, including envelope glycoproteins spike (S), envelope (E), membrane (M) and nucleocapsid (N). In addition to the genes encoding structural proteins, there are accessory genes that are species-specific and dispensable for virus replication. Here, we compare prototypical and representative strains of four coronavirus genera: feline infectious peritonitis virus (FIPV), Rhinolophus bat coronavirus HKU2, severe acute respiratory syndrome coronavirus (SARS-CoV) strains GD02 and SZ3 from humans infected during the early phase of the SARS epidemic and from civets, respectively, SARS-CoV strain hTor02 from humans infected during the middle and late phases of the SARS epidemic, bat SARS-related coronavirus (SARSr-CoV) strain WIV1, Middle East respiratory syndrome coronavirus (MERS-CoV), mouse hepatitis virus (MHV), infectious bronchitis virus (IBV) and bulbul coronavirus HKU11.
---

Fig. 2. Animal origins of human coronaviruses. Severe acute respiratory syndrome coronavirus (SARS-CoV) is a new coronavirus that emerged through recombination of bat SARS-related coronaviruses (SARSr-CoVs). The recombined virus infected civets and humans and adapted to these hosts before causing the SARS epidemic,. Middle East respiratory syndrome coronavirus (MERS-CoV) likely spilled over from bats to dromedary camels at least 30 years ago and since then has been prevalent in dromedary camels. HCoV-229E and HCoV-NL63 usually cause mild infections in immunocompetent humans. Progenitors of these viruses have recently been found in African bats,, and the camelids are likely intermediate hosts of HCoV-229E,. HCoV-OC43 and HKU1, both of which are also mostly harmless in humans, likely originated in rodents. Recently, swine acute diarrhoea syndrome (SADS) emerged in piglets. This disease is caused by a novel strain of Rhinolophus bat coronavirus HKU2, named SADS coronavirus (SADS-CoV); there is no evidence of infection in humans. Solid arrows indicate confirmed data. Broken arrows indicate potential interspecies transmission. Black arrows indicate infection in the intermediate animals, yellow arrows indicate a mild infection in humans, and red arrows indicate a severe infection in humans or animals.
---

Fig. 3. Phylogenetic relationships in the Coronavirinae subfamily. The highly human-pathogenic coronaviruses belong to the subfamily Coronavirinae from the family Coronaviridae. The viruses in this subfamily group into four genera (prototype or representative strains shown): Alphacoronavirus (purple), Betacoronavirus (pink), Gammacoronavirus (green) and Deltacoronavirus (blue). Classic subgroup clusters are labelled 1a and 1b for the alphacoronaviruses and 2a–2d for the betacoronaviruses. The tree is based on published trees of Coronavirinae, and reconstructed with sequences of the complete RNA-dependent RNA polymerase-coding region of the representative coronaviruses (maximum likelihood method under the GTR + I + Γ model of nucleotide substitution as implemented in PhyML, version 3.1 (ref.)). Only nodes with bootstrap support above 70% are shown. IBV, infectious bronchitis virus; MERS-CoV, Middle East respiratory syndrome coronavirus; MHV, mouse hepatitis virus; PEDV, porcine enteric diarrhoea virus; SARS-CoV, severe acute respiratory syndrome coronavirus; SARSr-CoV, SARS-related coronavirus.
---

Fig. 4. Phylogenetic analysis of SARSr-CoVs and MERSr-CoVs. a | The figure shows a simplified phylogenetic tree of severe acute respiratory syndrome-related coronaviruses (SARSr-CoVs) from bats. SARSr-CoVs cluster into three lineages, L1–L3, and human severe acute respiratory syndrome coronaviruses (SARS-CoVs) embed in L1. Two individual SARSr-CoVs do not cluster into these lineages: YN, a virus isolated from Yunnan province, China, and BG, a virus from Bulgaria, Europe. The tree is based on published trees, and reconstructed using sequences of the complete RNA-dependent RNA polymerase-coding region (maximum likelihood method under the GTR + I + Γ model of nucleotide substitution as implemented in PhyML, version 3.1 (ref.)).The strain Zhejiang2013 (GenBank No. KF636752) was used as a root. b | By contrast, Middle East respiratory syndrome-related coronaviruses (MERSr-CoVs) form two major viral lineages, L1 and L2. L1 is found in humans and camels, and L2 is found only in camels. Two small clusters, B1 (bat 1) and B2, and one single virus, SA, from South Africa, were found in bats. The phylogenetic tree of MERSr-CoVs is based on a published trees, and reconstructed using full-genome alignment of all coding regions using the same method as above. HKU4-1 (EF065505) and HKU5-1 (EF065509), two 2c betacoronaviruses, served as the root of the tree. Detailed phylogenetic trees and grouping information can be found in Supplementary Fig. S1. MERS-CoV, Middle East respiratory syndrome coronavirus.
---

Fig. 5. Variable regions in different SARS-CoV and bat SARSr-CoV isolates. Variability and thus species adaptation majorly affect three severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-related coronavirus (SARSr-CoV) proteins: the spike protein (S) (both the S1 amino-terminal domain (S1-NTD) and the S1 receptor-binding domain (S1-RBD) show variability), ORF3 (3a and 3b) and ORF8 (8a and b). SARS-CoV GD02 and hTor02 represent strains that were isolated from patients during the early, and middle or late phase of the SARS epidemic in 2002–2003, respectively; SARS-CoV CZ3 is a representative of strains isolated from civets in 2003 and 2004 (refs,). All bat SARSr-CoVs, except HKU3 and Rp3, were discovered in Yunnan province during 2011–2015. On the basis of deletions in the RBD, bat SARSr-CoVs can be divided into two clades. Those without a deletion and thus an identical size in S1 to SARS-CoV can be further divided into four genotypes: genotype 1, represented by WIV16, is highly similar to SARS-CoV in both the NTD and the RBD; genotype 2, represented by WIV1, differs in NTD from SARS-CoV; genotype 3, represented by Rs4231, differs in RBD from SARS-CoV; and genotype 4, represented by SHC014 and Rs4084, differs in both NTD and RBD from SARS-CoV. The differences in S influence species-specific receptor binding, whereas differences in the accessory proteins, including potentially the newly discovered ORFX (X), mainly affect immune responses and viral immune evasion. Adapted from ref., CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
---

Fig. 6. Receptor recognition by SARS-CoV and MERS-CoV. a | Severe acute respiratory syndrome coronavirus (SARS-CoV) uses its receptor-binding domain (RBD) (as shown in the structure of strain hTor02, containing core structure (cyan) and receptor-binding motif (RBM; magenta)) to bind human angiotensin-converting enzyme 2 (ACE2; green; Protein Data Bank ID: 2AJF). ACE2 is a peptidase with zinc (blue) in its active centre. b | Several residues in the host and viral receptor, as well as two salt bridges that stabilize the structure (dotted lines) and form two binding hot spots, are crucial for binding of the severe acute respiratory syndrome (SARS) epidemic strain hTor02. Hydrophobic residues surrounding the two salt bridges are present in the structure but are not shown in the figure. c | By contrast, the SARS-related coronavirus (SARSr-CoV) strain bWIV1, which was isolated from bats and can infect both civet and human cells, differs in residues 442, 472 and 487. The mutation from threonine to asparagine in residue 487 introduces a polar side chain and is predicted to interfere with binding at hot spot 353. The model shown here was built on the basis of the structure of hTor02 RBD complexed with human ACE2 (Protein Data Bank ID: 2AJF), in which residues 442, 472 and 487 were mutated from those in strain hTor02 to those in strain bWIV1. d | The bat SARSr-CoV strain bRsSHC014 can also infect human and civet cells; it carries an alanine in position 487, and the short side chain of this residue does not support the structure of hot spot 353. The model was built on the basis of the structure of cOptimize RBD complexed with human ACE2 (Protein Data Bank ID: 3SCJ), in which residues 442, 480 and 487 were mutated from those in strain cOptimize to those in strain bWIV1. e | The Middle East respiratory syndrome coronavirus (MERS-CoV) RBD (core structure in cyan and RBM in magenta) binds human dipeptidyl peptidase 4 (DPP4; green; Protein Data Bank ID: 4KR0). Structure figures were made using PyMOL. Modelled mutations in panels c and d were performed using Coot. Panels a–d are adapted from ref.: this research was originally published in The Journal of Biological Chemistry. Wu, K. L., Peng, G. Q., Wilken, M., Geraghty, R. J. & Li, F. Mechanisms of host receptor adaptation by severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2012; 287:8904–8911. © American Society for Biochemistry and Molecular Biology.
--- Full read: pubmed.ncbi.nlm.nih.gov/30531947/?from_term=coronavirus&from_pos=8
|
|